

Sie möchten uns unterstützen?
Die Selbsthilfegruppe finanziert sich durch Spenden und freie Zuwendungen. Mit Ihrer Spende helfen Sie, unsere Arbeit in der Plasmozytom / Multiples Myelom Selbsthilfe zu unterstützen.
Das MM ist eine Erkrankung, die von Patient zu Patient ganz unterschiedlich verläuft. Aus diesem Grund ist die ausführliche und individuelle Beratung jedes einzelnen Myelompatienten durch einen in der Diagnostik und Therapie der Erkrankung erfahrenen Arzt unverzichtbar. Bei einigen Myelompatienten bleibt die Erkrankung über viele Jahre hinweg vergleichsweise inaktiv und muss nicht behandelt werden (sogenanntes asymptomatisches oder "smoldering" (= schwelendes) Myelom). Bei anderen Patienten zeigt die Erkrankung einen äußerst aggressiven Verlauf und führt binnen weniger Monate trotz intensiver Therapiebemühungen zum Tod. Sehr wahrscheinlich gibt es nicht eine Erkrankung "Multiples Myelom", sondern viele Krankheitsvarianten mit ganz unterschiedlichen biologischen Eigenschaften. So gibt es beispielsweise immer wieder Myelompatienten, bei denen keine oder nur ganz diskrete Knochenveränderungen nachweisbar sind. Andere Patienten weisen bevorzugt Krankheitsherde außerhalb des Knochenmarkes auf ("extramedulläre" Tumormanifestationen). Bei wieder anderen Patienten produzieren die Plasmazellen kein Eiweiß ("asekretorisches" Myelom, ca. 2-3% aller Betroffenen).
Man ist heute in der Lage, den Krankheitsverlauf eines Patienten anhand bestimmter Krankheitsmerkmale (Prognosemarker) grob abzuschätzen. Hierzu zählen bestimmte Laborwerte, einige Veränderungen am Erbgut der Tumorzelle (genetische Veränderungen) sowie eine Reihe von "klinischen" Eigenschaften der Erkrankung (z.B. die Qualität und Dauer eines Therapieansprechens). Es ist jedoch entscheidend zu verstehen, dass die Bedeutung jedes einzelnen Prognosemarkers an großen Patientengruppen untersucht wurde und dieser daher nur eine beschränkte Aussagekraft für den einzelnen Patienten besitzt. Man spricht in diesem Zusammenhang von "Mittelwerten" bestimmter Messgrößen (z.B. der Überlebenszeit), die aus den Werten (hier beispielhaft: Überlebenszeit) aller Patienten einer Gruppe errechnet wurden. Bei einem individuellen Patienten kann der Wert der Messgröße erheblich vom Mittelwert abweichen, im Fall der Überlebenszeit also deutlich länger, aber auch deutlich kürzer sein. Da Prognosemarker die Auswahl der Behandlung eines Myelompatienten mit beeinflussen können, sollten die entsprechenden Befunde zwischen Patient und Arzt ausführlich besprochen werden. Die heute wichtigsten Prognosemarker sind nachfolgend kurz beschrieben.
Der ß2-Mikroglobulinwert (sprich: Beta-2-Mikroglobulinwert) ist beim MM ein Maß für die Tumormasse (je höher der Wert, umso höher die Tumorzellgesamtanzahl). Auch bei einer Nierenfunktionsstörung, die bei Patienten mit MM häufig vorliegt, sind erhöhte ß2-Mikroglobulinwerte zu finden, da dieses Eiweißmolekül über die Niere ausgeschieden wird. In vielen Studien war ein erhöhter ß2-Mikroglobulinwert (unabhängig von der Nierenfunktion) mit einem kürzeren Überleben der Patienten vergesellschaftet. In Kombination aus ß2-Mikroglobulin und Serumalbuminwert, der einen Hinweis auf die Zusammensetzung der Eiweiße im Blut gibt, berechnet sich das Stadium nach dem "International Staging System" (ISS). Der ß2-Mikroglobulin- und Serumalbuminspiegel werden in der Regel nicht als Verlaufsparameter verwendet, sondern einmalig bei Erstdiagnose bestimmt.
eine Erhöhung des LDH-Wertes tritt immer auf, wenn Zellen (nicht nur Myelomzellen) zugrunde gehen. Somit muss eine LDH-Erhöhung bei einem Myelompatienten nicht zwangsläufig auf die Myelomerkrankung zurückzuführen sein. Ist die LDH-Erhöhung jedoch durch die Erkrankung bedingt, muss von einer sehr aktiven Erkrankung ausgegangen werden. Eine Normalisierung des LDH-Wertes zeigt ein Ansprechen auf eine bestimmte Therapie an.
Unter Deletion versteht man den Verlust von Erbsubstanz im Sinne eines Verlustes eines ganzen Chromosoms oder eines Chromosomenbruchstückes (Abbildungen 6 und 7).
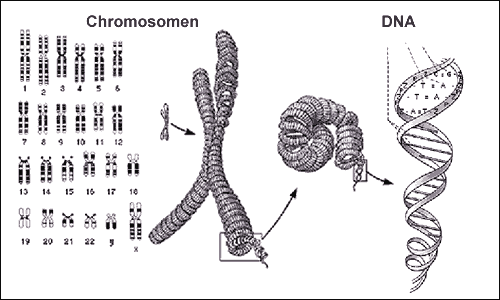
Die Erbinformation des Menschen ist in seiner DNA verschlüsselt. Vor der Zellteilung wird die DNA in Form von Chromosomen verpackt. Eine normale Zelle verfügt über 46 Chromosomen (Chromosomen Nr. 1-22 in zweifacher Ausführung und zwei Geschlechtschromosomen – XX bei der Frau und XY beim Mann).
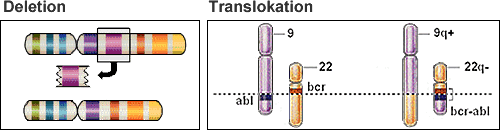
Schema einer Deletion (oben links) sowie einer Translokation (oben rechts) als häufige Veränderungen am Erbgut von Myelomzellen
Unter Deletion versteht man den Verlust von Erbsubstanz. Dabei kann entweder ein ganzes Chromosom oder ein Chromosomenabschnitt verloren gehen. (Abbildungen 6 und 7). Deletionen des kurzen Arms von Chromosom 17 (17p) sind bei Erstdiagnose relativ selten zu finden (ca. 10% aller Patienten). Bei fortgeschrittenen Erkrankungen (nach mehreren Rückfällen) nimmt die Rate an 17p-Verlusten deutlich zu. Durch diese genetische Veränderung wird das Gen TP53 , welches das Krebswachstum hemmt, in seiner Funktion gestört. In der Regel sprechen Patienten mit einer Deletion 17p entweder gar nicht auf die Therapie an oder erleiden einen schnellen Krankheitsrückfall. Ein besonders hohes Risiko besteht, wenn zusätzlich zu der Deletion noch eine Mutation des verbliebenen Gens auftritt, sodass die hemmende Funktion komplett entfällt. Ob diese Patienten von neuen Immuntherapien, z.B. einer CAR-T-Zelltherapie oder bispezifischen Antikörpern, profitieren bleibt abzuwarten.
Bei einer Translokation kommt es zu einem Austausch von Erbsubstanz zwischen zwei Chromosomen (siehe Abb. 7 rechts), im Falle der Translokation t(4;14) zwischen einem Chromosom 4 und einem Chromosom 14. Durch eine Translokation kann die Aktivität eines oder mehrere Krebsgene gesteigert werden. Etwa 15% aller Myelompatienten tragen eine t(4;14) in ihren Tumorzellen. Nicht alle Patienten mit einer t(4;14) leiden unter einem frühen Krankheitsrückfall. Allerdings ist es noch nicht routinemäßig möglich, diese Patienten zu identifizieren.
Der Zugewinn einer Region des Chromosoms 21 (Arm q, Bande 21 = 1q21) ist die häufigste prognostisch ungünstige Veränderung beim Multiplen Myelom. Sie tritt bei bis zu 40% der neudiagnostizierten Patienten auf. Es kann auch zu mehreren Zugewinnen von 1q21, einer sogenannten Amplifikation, kommen. Patienten mit einem einfachen Zugewinn oder einer Amplifikation unterscheiden sich jedoch nicht in ihrer Prognose.
Neuere Untersuchungen haben gezeigt, dass eine Deletion von Chromosom 13 nicht mit einem frühen Krankheitsrückfall assoziiert ist. Diese Deletion wird daher nicht mehr zur Prognosebeurteilung verwendet.
Die prognostische Bedeutung weiterer Aberrationen (z.B. Trisomie 1q) wird kontrovers diskutiert oder ist ungeklärt. Eine wichtige Erkenntnis ist,
dass die Anzahl der Veränderungen mit prognostischer Bedeutung, die gleichzeitig auftreten, eine wichtige Rolle spielt: je mehr prognostisch ungünstige Varianten vorliegen, desto früher kommt es in der Regel zu einem Krankheitsrückfall.
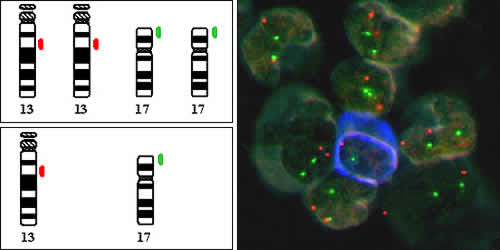
FISH-Technik zum Nachweis genetischer Veränderungen. Eingesetzt werden Gensonden, an die rote und grüne Leuchtfarbstoffe gebunden sind. In diesem Beispiel wurden Myelomzellen mit einem speziellen Farbstoff blau angefärbt, um sie von normalen Zellen zu unterscheiden. In der abgebildeten Myelomzelle zeigt sich, im Gegensatz zu allen anderen (normalen) Blutzellen, das Fehlen jeweils einer Gensonden-Kopie, was in diesem Fall Deletionen von 13q und 17p anzeigt.
Stadium nach Salmon & Durie: die Stadieneinteilung nach Salmon & Durie wurde im Jahr 1975 eingeführt und basiert auf einer Reihe einfach bestimmbarer Messgrößen (Paraproteinmenge, Kalzium- und Hämoglobinwert, Grad der radiologischen Knochenveränderungen; Abbildung 9).
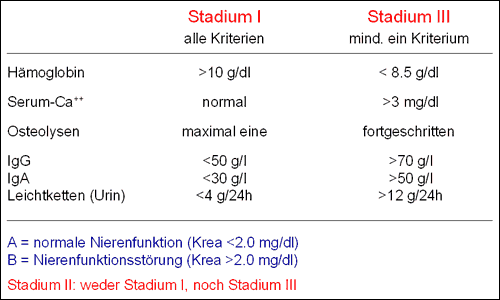
Unterschieden werden die drei Stadien I, II und III. Zusätzlich wird eine ein bestimmtes Maß überschreitende Nierenfunktionseinschränkung mit dem Kürzel "B" gekennzeichnet, während bei weitgehend normaler Nierenfunktion ein "A"-Stadium vorliegt. Das Stadium nach Salmon & Durie gilt als orientierendes Maß für die Tumormasse, ist aber kein zuverlässiger Prognosemarker. Der bei weitem größte Teil der Patienten befindet sich bei Diagnosestellung im Stadium III der Erkrankung.
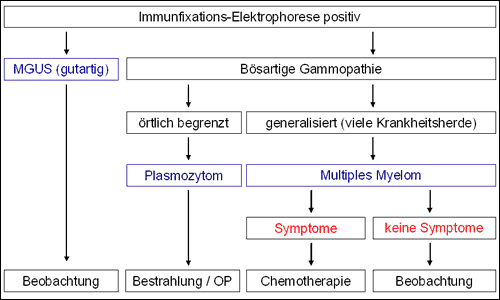
Für die Klinik entscheidender als die reine Stadieneinteilung ist heute die Unterscheidung zwischen gutartiger (MGUS) bzw. bösartiger (Plasmozytom/MM), lokalisierter (Plasmozytom) bzw. generalisierter (MM) sowie symptomatischer bzw. asymptomatischer Erkrankung, da sich hieraus Einflüsse auf die Therapieentscheidungen (keine Therapie; örtlich begrenzte Behandlung, wie etwa Strahlentherapie; Chemotherapie) ergeben (Abbildungen 10 und 11).
Als symptomatisches MM mit Notwendigkeit einer Behandlung wird eine Erkrankung bezeichnet, die mit einer Kalziumerhöhung im Blut (Hyperkalziämie), einer signifikanten Störung der Bildung roter Blutkörperchen (Anämie), einer deutlichen Nierenfunktionsverschlechterung oder einer Knochenproblematik (Osteolysen) einhergeht (Abbildung 11).
Da sich gezeigt hatte, dass aber auch Patienten mit asymptomatischer Erkrankung bei Vorliegen bestimmter Risikofaktoren von einer frühen Therapieeinleitung profitieren können, wurde kürzlich zu den o.g. Symptomen noch so genannte "Myelom-definierende Ereignisse" hinzugefügt. Diese sind das Vorhandensein einer, einen bestimmten Grenzwert überschreitenden Leichtkettenkonzentration im Serum, das Vorhandensein von mehr als einer fokalen Veränderung in der MRT-Untersuchung sowie ein Nachweis einer Vermehrung von monoklonalen (kranken) Plasmazellen im Knochenmark von 60% oder mehr. Diese neuen Kriterien müssen aber durch einen in der Myelomdiagnostik und -behandlung erfahrenen Arzt beurteilt und ihrer Bedeutung eingeschätzt werden, da zumindest bei den erstgenannten Parametern auch langfristige Verläufe ohne erhöhtes Progressionsrisiko gefunden werden können.

Zeigen sich bei der Knochenmarkuntersuchung vermehrt sehr unreife Plasmazellen (Plasmablasten), muss von einem aggressiven Krankheitsverlauf und einer schlechteren Prognose ausgegangen werden.
Spricht ein Patient auf eine chemotherapeutische Behandlung gar nicht an, oder tritt ein Rückfall der Erkrankung sehr rasch nach Abschluss der Therapie auf, muss von einer gewissen Widerstandsfähigkeit der Tumorzellen gegenüber Zellgiften (Chemotherapie-Substanzen) ausgegangen werden, was die Behandlung der Erkrankung deutlich erschwert. Welche Bedeutung ein unzureichendes Ansprechen der Tumorzellen auf klassische Zellgifte im Hinblick auf die Effektivität neuer Substanzen, wie Bortezomib, Thalidomid oder Lenalidomid hat, ist derzeit noch ungeklärt.
(Nachfragen an Dr. Hillengaß)
Können Plasmazellen in der Blutbahn nachgewiesen werden und machen sie dort mehr als 20% aller weißen Blutkörperchen aus, spricht man von einer Plasmazell-Leukämie. Eine Plasmazell-Leukämie ist mit einer sehr schlechten Prognose vergesellschaftet.
Autoren:
PD Dr. med. Peter Liebisch, aktualisiert und überarbeitet Ende 2015 von Prof. Dr. med. Jens Hillengaß (siehe Einleitung)
Eine Studie der Uniklinik Heidelberg zur prognostischen Asusagekraft chromosomaler Veränderungen: